Stage 4: Intervention design
In this tutorial, we’ll walk through how to use the CASCADE model trained in stage 2 to perform targeted intervention design, using K562-to-erythroid differentiation as an example.
[1]:
import anndata as ad
import numpy as np
import pandas as pd
import scanpy as sc
import seaborn as sns
from scipy.sparse import csr_matrix
from cascade.data import (
configure_dataset,
encode_regime,
get_all_targets,
get_configuration,
)
from cascade.model import CASCADE, IntervDesign
from cascade.plot import plot_design_error_curve, plot_design_scores, set_figure_params
[2]:
set_figure_params()
Prepare source data
First, we will extract the source state, i.e., unperturbed K562 cells from our preprocessed data:
[3]:
adata = sc.read_h5ad("adata.h5ad")
adata
[3]:
AnnData object with n_obs × n_vars = 86744 × 1064
obs: 'guide_id', 'gemgroup', 'ncounts', 'knockup'
var: 'perturbed', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'selected'
uns: '__CASCADE__', 'hvg', 'log1p', 'pca'
obsm: 'X_pca', 'covariate'
varm: 'PCs'
layers: 'counts', 'interv'
[4]:
source = adata[adata.obs["knockup"] == ""].copy()
source
[4]:
AnnData object with n_obs × n_vars = 11855 × 1064
obs: 'guide_id', 'gemgroup', 'ncounts', 'knockup'
var: 'perturbed', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'selected'
uns: '__CASCADE__', 'hvg', 'log1p', 'pca'
obsm: 'X_pca', 'covariate'
varm: 'PCs'
layers: 'counts', 'interv'
Prepare target data
Next, we use an erythroid scRNA-seq dataset from Xu, et al. (2022) as the target state. The data file can be downloaded from here:
[5]:
target = sc.read_h5ad("Xu-2022.h5ad")
target
[5]:
AnnData object with n_obs × n_vars = 74460 × 27069
obs: 'Clusters', 'batch', 'stage', 'ncounts'
Some genes in our source dataset could be missing from the target data, so we manually expand the target data with zero-paddings:
[6]:
common_vars = [i for i in adata.var_names if i in target.var_names]
missing_vars = [i for i in adata.var_names if i not in target.var_names]
len(common_vars), len(missing_vars)
[6]:
(930, 134)
[7]:
empty = ad.AnnData(
X=csr_matrix((target.n_obs, len(missing_vars)), dtype=target.X.dtype),
obs=target.obs,
var=pd.DataFrame(index=missing_vars),
)
target = ad.concat([target, empty], axis=1, merge="first")
target
[7]:
AnnData object with n_obs × n_vars = 74460 × 27203
obs: 'Clusters', 'batch', 'stage', 'ncounts'
Similar to the training data, we backup the raw counts and log-normalize the dataset.
[8]:
target.layers["counts"] = target.X.copy()
sc.pp.normalize_total(target, target_sum=1e4)
sc.pp.log1p(target)
We’d also need to configure the target dataset. Of note, the target data configuration should provide the same covariate as the training data. However, it obviously does not have batch info compatible with the training data.
Here we are using a rough approach of simply taking the covariate of random training cells. Another possibility is to take the covariate of training cells most similar to the target cells, based on the expression levels of housekeeping genes.
[9]:
target.obsm["covariate"] = source.obsm["covariate"][
np.random.choice(source.n_obs, target.n_obs)
]
[10]:
configure_dataset(
target, use_covariate="covariate", use_size="ncounts", use_layer="counts"
)
get_configuration(target)
[10]:
{'regime': None,
'covariate': 'covariate',
'size': 'ncounts',
'weight': None,
'layer': 'counts'}
Define gene weights
Given that the source and target data were produced with different experimental protocols in different studies, batch effect would be a substantial problem. To mitigate this issue, we give higher weight to known erythroid markers when comparing counterfactual states with the target state during intervention design, which helps avoid learning batch effect between source and target.
[11]:
markers = [
"AHSP",
"ALAS2",
"ALDOA",
"BLVRB",
"BPGM",
"CLIC1",
"ENO1",
"GYPA",
"GYPB",
"HAMP",
"HBA1",
"HBA2",
"HBB",
"HBD",
"HBE1",
"HBG1",
"HBG2",
"HBZ",
"HEMGN",
"LDHA",
"PRDX1",
"PRDX2",
"SLC25A37",
"SLC4A1",
"SMIM1",
]
assert not set(markers) - set(adata.var_names)
assert not set(markers) - set(target.var_names)
len(markers)
[11]:
25
[12]:
non_markers = [i for i in common_vars if i not in markers]
len(non_markers)
[12]:
905
[13]:
target.var["weight"] = 0.0
target.var.loc[non_markers, "weight"] = len(common_vars) / len(non_markers) / 2
target.var.loc[markers, "weight"] = len(common_vars) / len(markers) / 2
[14]:
target.var.loc[non_markers, "weight"].head()
[14]:
SAMD11 0.513812
HES4 0.513812
ISG15 0.513812
RNF223 0.513812
TNFRSF4 0.513812
Name: weight, dtype: float64
[15]:
target.var.loc[markers, "weight"].head()
[15]:
AHSP 18.6
ALAS2 18.6
ALDOA 18.6
BLVRB 18.6
BPGM 18.6
Name: weight, dtype: float64
In this case we scaled the gene weights so that the total weight of marker genes equals and the total weight of non-marker genes, and genes that were zero-padded in the target data were given zero weight, so they will not bias the result.
Specify candidate genes
We can also provide a candidate gene pool, e.g., we’ll just use genes perturbed in the CRISPRa dataset that show higher expression in the target cells:
[16]:
all_targets = sorted(get_all_targets(adata, "knockup"))
target_mask = (
target[:, all_targets].to_df().mean() > adata[:, all_targets].to_df().mean()
)
candidates = target_mask.index[target_mask].to_list()
candidates
[16]:
['ARRDC3',
'ATL1',
'BPGM',
'CDKN1A',
'CDKN1B',
'CDKN1C',
'CEBPA',
'CEBPB',
'COL1A1',
'CSRNP1',
'EGR1',
'ETS2',
'FOSB',
'FOXF1',
'HNF4A',
'HOXA13',
'IER5L',
'IGDCC3',
'IRF1',
'JUN',
'KLF1',
'MAML2',
'MAP2K3',
'MAP2K6',
'MAP3K21',
'MAP4K5',
'MEIS1',
'MIDEAS',
'PRDM1',
'PRTG',
'PTPN13',
'RUNX1T1',
'SGK1',
'SLC38A2',
'SLC4A1',
'SNAI1',
'SPI1',
'TBX3',
'TGFBR2',
'TSC22D1',
'ZBTB1',
'ZBTB10']
Run intervention design
(Estimated time: 10 min – 20 min, depending on computation device)
Finally, we can load our trained model for intervention design:
[17]:
cascade = CASCADE.load("tune.pt")
For the sake of speed, we will subsample both the source and target data to 5,000 cells.
[18]:
sc.pp.subsample(source, n_obs=5000)
sc.pp.subsample(target, n_obs=5000)
CASCADE model provides a dedicated design method for intervention design, which uses differentiable optimization to optimize interventions that produces effects more similar to the target state.
We’ll need to pass the source and target datasets, along with a candidate gene pool, a maximal combination order (design_size=1 for designing single-gene perturbations), as well as the target gene weight we just assigned.
[19]:
scores, design = cascade.design(
source, target, pool=candidates, design_size=1, target_weight="weight"
)
17:32:38.143 | WARNING | 3810563:model:align_vars - 26139 variables are not in the `scaffold` and will thus be ignored.
17:32:38.151 | INFO | 3810563:utils:autodevice - Using GPU [1] as computation device.
╭────────────────────────────── cascade-reg ──────────────────────────────╮ │ │ │ Training on 1064 variables with 12294 scaffold edges and 5000 samples │ │ │ ╰──────────────────────────────── v0.4.0 ─────────────────────────────────╯
17:32:38.325 | INFO | 3810563:core:fit_stage - Number of topological generations: [68, 88, 70, 101]
| Name | Type | Params | Mode
------------------------------------------------------
0 | scaffold | Edgewise | 49.2 K | eval
1 | sparse | L1 | 0 | eval
2 | acyc | SpecNorm | 0 | eval
3 | kernel | RBF | 0 | eval
4 | latent | EmbLatent | 6.3 K | eval
5 | lik | NegBin | 0 | eval
6 | func | Func | 7.9 M | eval
7 | design | IntervDesign | 8.6 K | train
| other params | n/a | 8.5 K | n/a
------------------------------------------------------
10.7 K Trainable params
8.0 M Non-trainable params
8.0 M Total params
31.955 Total estimated model params size (MB)
1 Modules in train mode
16 Modules in eval mode
Restoring best model: log_dir/design/lightning_logs/version_0/checkpoints/epoch=8-step=1200.ckpt.
The design method returns two objects:
The
scoresobject is a data frame containing scores of candidate genes (or gene combinations ifdesign_sizewas larger than 1).The
designobject is an IntervDesign module that contains both the scores and optimized interventional scales and biases for the designed interventions, which can also be saved and loaded just like the CASCADE model.
[20]:
scores.to_csv("design.csv")
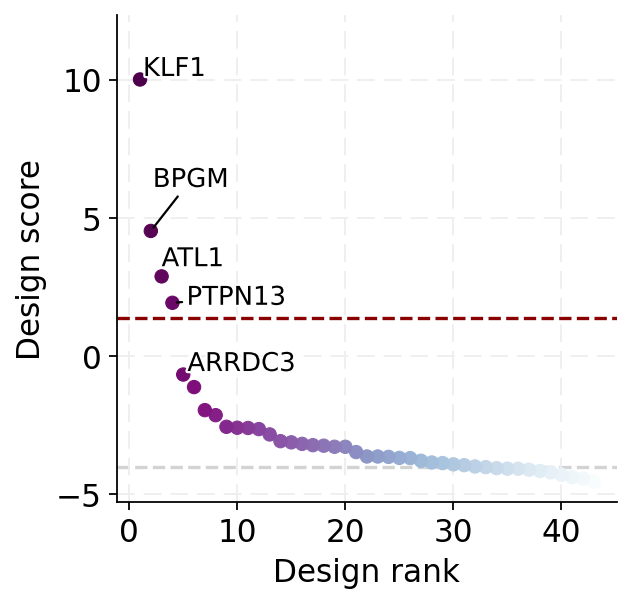
scores.head()
[20]:
| score | |
|---|---|
| KLF1 | 10.006369 |
| BPGM | 4.527291 |
| ATL1 | 2.883361 |
| PTPN13 | 1.928048 |
| ARRDC3 | -0.666598 |
[21]:
design.save("design.pt")
design = IntervDesign.load("design.pt")
The same can also be achieved using the command line interface with the following command.
cascade design -d source.h5ad -m tune.pt -t target.h5ad \
--pool candidates.txt -o design.csv -u design.pt \
--design-size 1 --target-weight weight [other options]
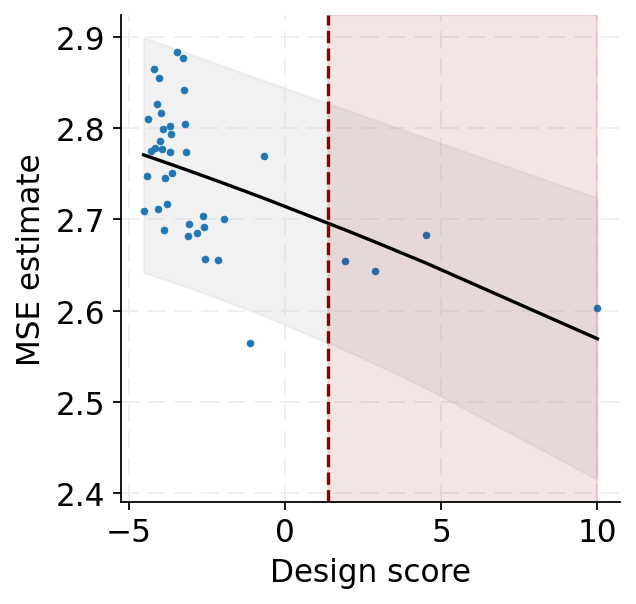
Determine design score cutoff
[22]:
curve, cutoff = cascade.design_error_curve(source, target, design, n_cells=5000)
18:02:56.569 | WARNING | 3810563:model:align_vars - 26139 variables are not in the `scaffold` and will thus be ignored.
/rd1/user/caozj/CASCADE/conda/lib/python3.11/site-packages/anndata/_core/anndata.py:1818: UserWarning: Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
utils.warn_names_duplicates("obs")
/rd1/user/caozj/CASCADE/conda/lib/python3.11/site-packages/anndata/utils.py:260: UserWarning: Suffix used (-[0-9]+) to deduplicate index values may make index values difficult to interpret. There values with a similar suffixes in the index. Consider using a different delimiter by passing `join={delimiter}`Example key collisions generated by the make_index_unique algorithm: ['ATTCTACAGCGTCAAG-1', 'CTTCTCTCATCGACGC-1', 'TACGGATGTGATGTCT-1', 'AGCCTAAAGAAACCTA-1', 'AACTCAGAGCTCCCAG-1']
warnings.warn(
18:03:00.430 | INFO | 3810563:core:predict_mode - Number of topological generations: [68, 88, 70, 101]
[23]:
plot_design_error_curve(curve, cutoff=cutoff)
[23]:
<Axes: xlabel='Design score', ylabel='MSE estimate'>
We can also visualize the scores with a scatter plot:
[24]:
plot_design_scores(scores, cutoff=cutoff)
[24]:
<Axes: xlabel='Design rank', ylabel='Design score'>
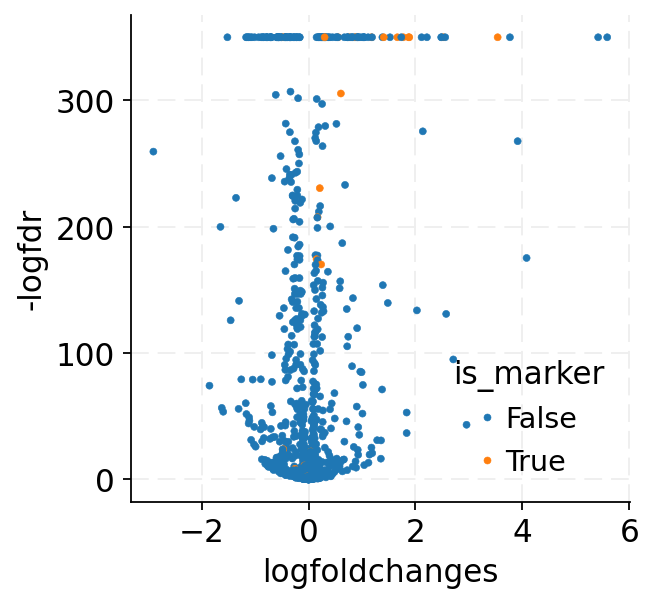
Verify design with counterfactual prediction
The designed perturbation can certainly be passed back to the counterfactual method to verify whether the target markers are up-regulated, just like what we did in stage 3.
One notable difference is that we need to specify the design argument of the counterfactual method, which tells the model to use the designed interventional scales and biases (instead of those from the training set) when making counterfactual predictions.
[25]:
source.obs["my_pert"] = "KLF1"
encode_regime(source, "ctfact", key="my_pert")
18:07:48.121 | WARNING | 3810563:data:encode_regime - Overwriting existing regime "ctfact".
[26]:
configure_dataset(source, use_regime="ctfact")
ctfact = cascade.counterfactual(source, design=design, sample=True)
18:07:48.188 | WARNING | 3810563:data:configure_dataset - Overwriting existing `regime` = "interv".
18:07:48.370 | INFO | 3810563:core:predict_mode - Number of topological generations: [68, 88, 70, 101]
[27]:
configure_dataset(source, use_regime="interv")
nil = cascade.counterfactual(source, design=design, sample=True)
18:07:56.710 | WARNING | 3810563:data:configure_dataset - Overwriting existing `regime` = "ctfact".
18:07:57.178 | INFO | 3810563:core:predict_mode - Number of topological generations: [68, 88, 70, 101]
[28]:
combined = ad.concat({"nil": nil, "ctfact": ctfact}, label="role", index_unique="-")
combined.X = np.log1p(combined.X * (1e4 / combined.obs[["ncounts"]].to_numpy()))
combined
[28]:
AnnData object with n_obs × n_vars = 10000 × 1064
obs: 'guide_id', 'gemgroup', 'ncounts', 'knockup', 'my_pert', 'role'
obsm: 'X_pca', 'covariate'
layers: 'counts', 'interv', 'ctfact', 'X_ctfact'
[29]:
sc.tl.rank_genes_groups(combined, "role", reference="nil", rankby_abs=True, pts=True)
de_df = sc.get.rank_genes_groups_df(combined, "ctfact").query("pct_nz_group > 0.05")
de_df["-logfdr"] = -np.log10(de_df["pvals_adj"]).clip(lower=-350)
de_df["is_marker"] = de_df["names"].isin(markers)
de_df.head()
/rd1/user/caozj/CASCADE/conda/lib/python3.11/site-packages/pandas/core/arraylike.py:399: RuntimeWarning: divide by zero encountered in log10
result = getattr(ufunc, method)(*inputs, **kwargs)
[29]:
| names | scores | logfoldchanges | pvals | pvals_adj | pct_nz_group | -logfdr | is_marker | |
|---|---|---|---|---|---|---|---|---|
| 0 | KLF1 | 492.331299 | 2.114285 | 0.0 | 0.0 | 1.0 | 350.0 | False |
| 1 | PNMT | 438.697296 | 2.480975 | 0.0 | 0.0 | 1.0 | 350.0 | False |
| 2 | TMSB10 | 366.669769 | 1.523769 | 0.0 | 0.0 | 1.0 | 350.0 | False |
| 3 | S100A10 | 247.035904 | 1.381347 | 0.0 | 0.0 | 1.0 | 350.0 | False |
| 4 | HBG2 | 236.489166 | 1.407301 | 0.0 | 0.0 | 1.0 | 350.0 | True |
[30]:
_ = sns.scatterplot(
data=de_df, x="logfoldchanges", y="-logfdr", hue="is_marker", edgecolor=None, s=10
)